Roadmap
Part I: Protein Structure
- Proteins, the PDB, and CASP
- AlphaFold 2 & RFdiffusion
- Representing protein structure
Part II: Small Molecules
- Molecular representations (SMILES, fingerprints, graphs)
- Binding prediction & its challenges
- AF3, Boltz-2 & the data vs. architecture question
Part I
Protein Structure
Proteins Are Molecular Machines
Proteins perform nearly every function in living cells:
- Catalyze reactions
- Transport molecules
- Enable movement
- Sense signals
- Fight infection
O(10,000) different proteins in a human cell
Sequence → Structure → Function
The amino acid sequence determines the 3D structure, which determines the function
ATP Synthase — The World's Smallest Rotary Motor
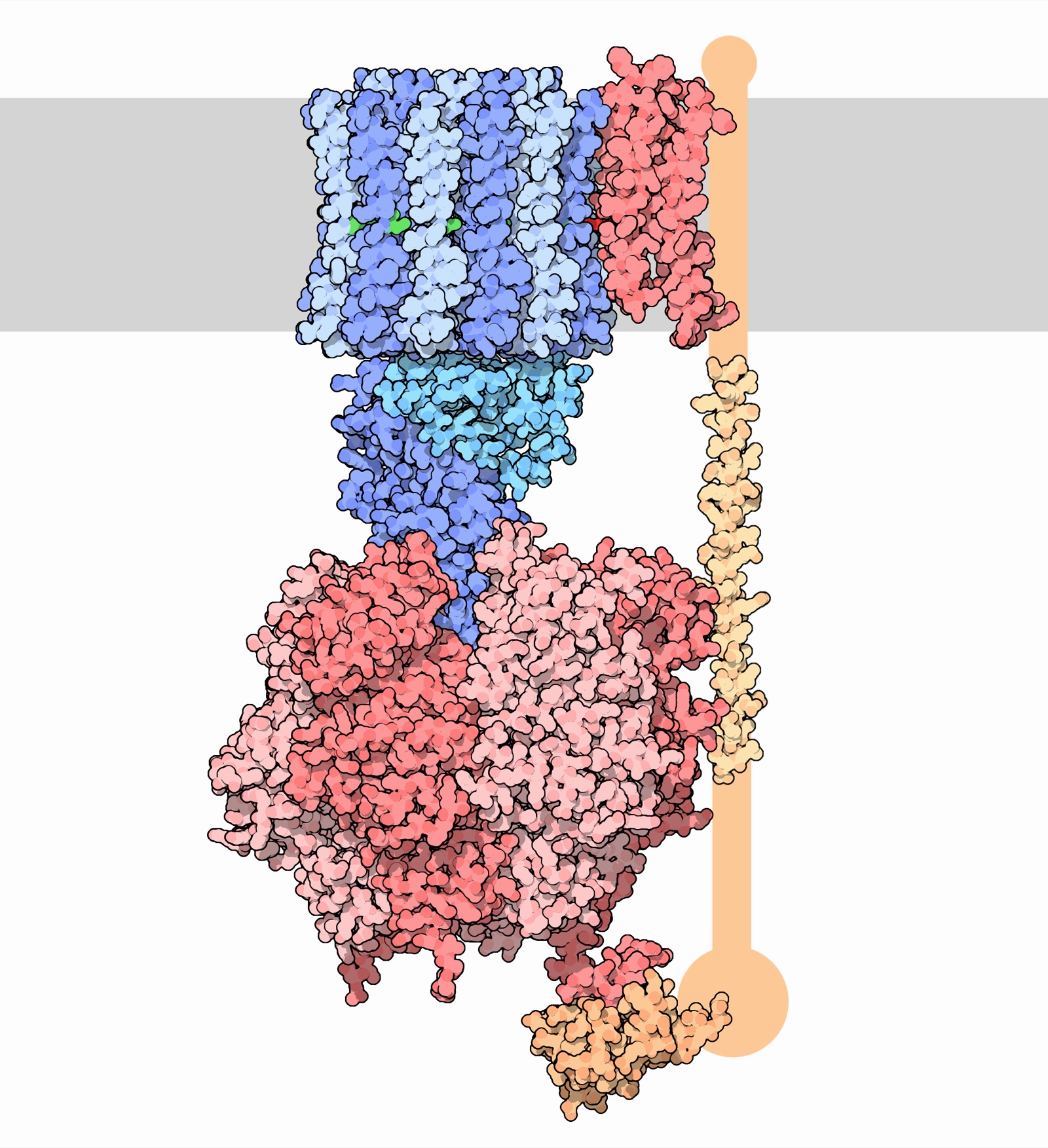
- Converts ADP + Pi → ATP using a proton gradient
- Rotates at ~9,000 RPM
- Produces ~100 kg of ATP per person per day
Image: David S. Goodsell, RCSB PDB
Viral Fusion Machinery
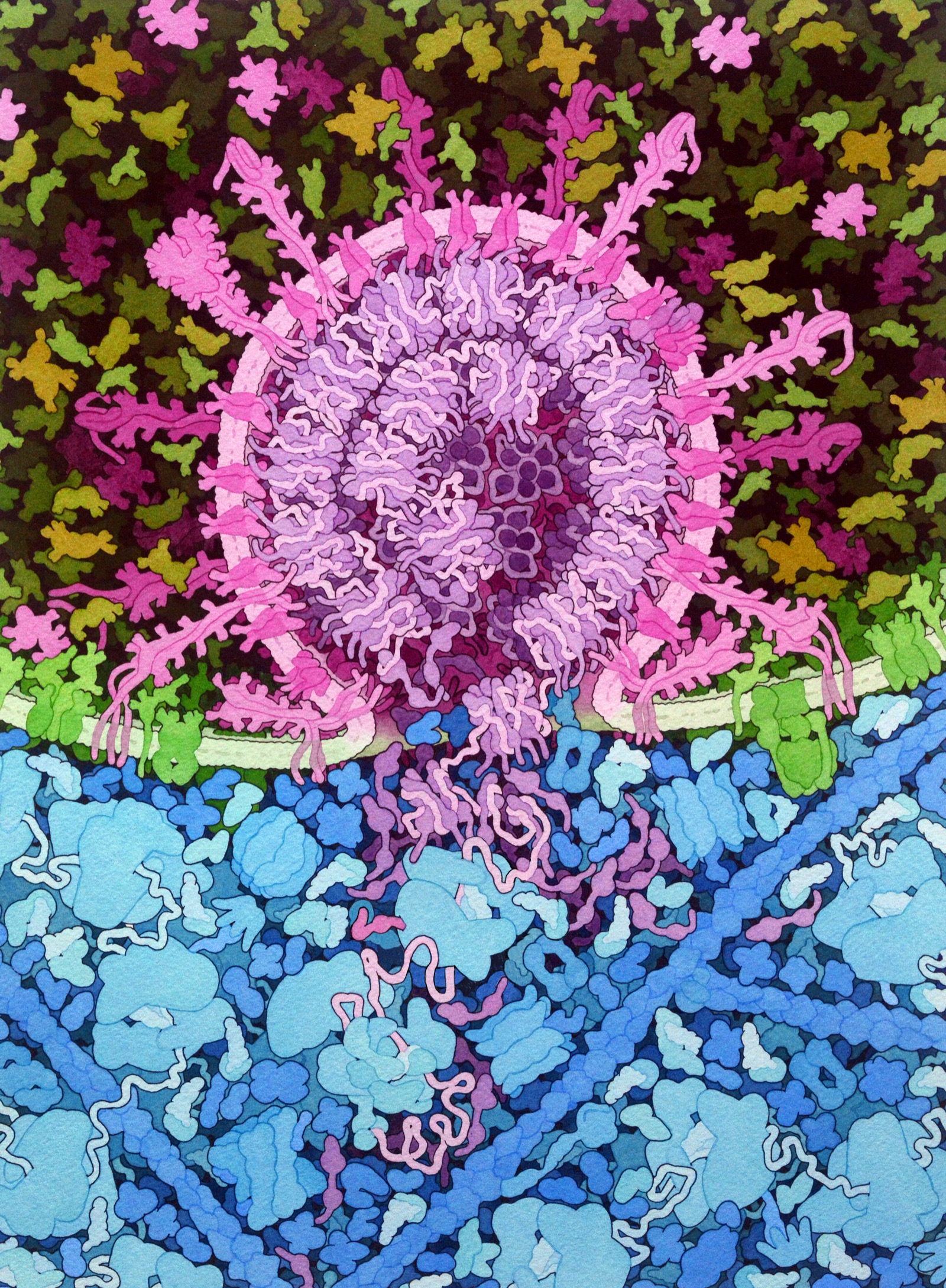
The SARS-CoV-2 spike protein binds ACE2 receptors and fuses viral and cell membranes
Understanding protein structure → vaccine design
(mRNA vaccines encode this protein)
Image: David S. Goodsell, RCSB PDB
Green Fluorescent Protein (GFP)
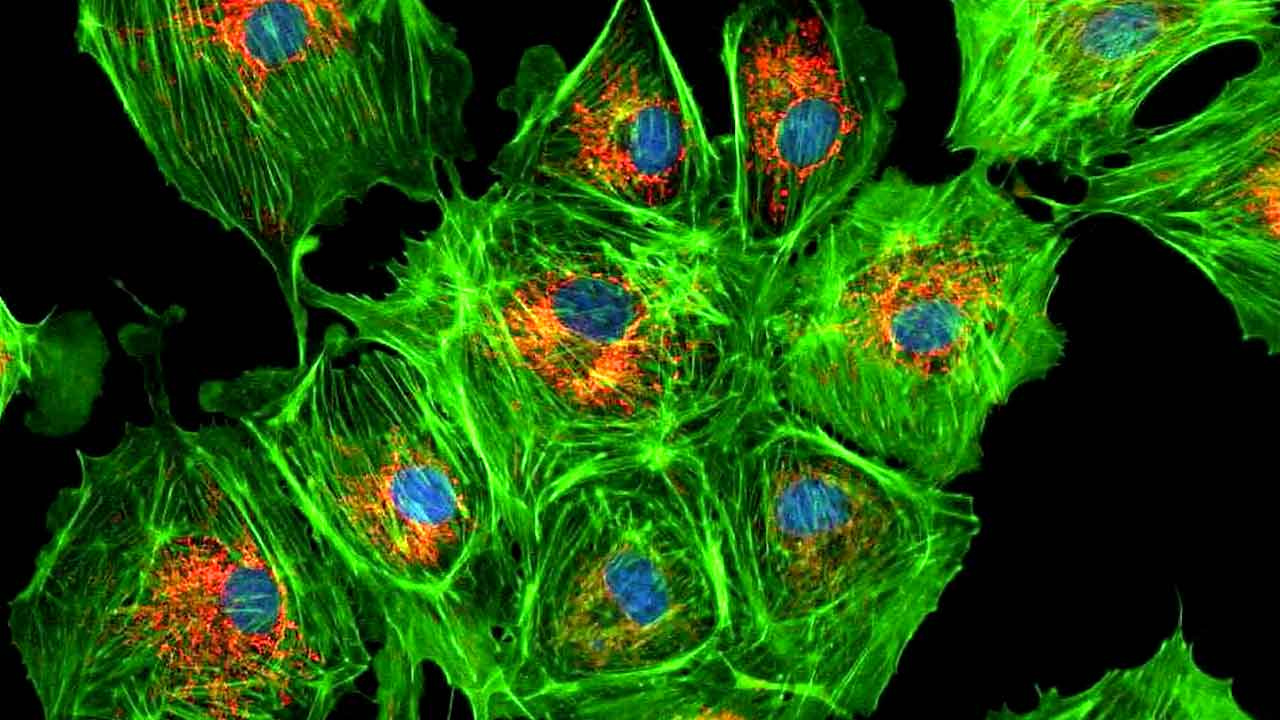
- Protein that literally glows — absorbs UV, emits green light
- Chromophore forms spontaneously from 3 amino acids
- Nobel Prize in Chemistry 2008
- Revolutionized biology: fuse GFP to any protein to track it in living cells
Amino Acids: The Building Blocks of Proteins
20 amino acids, each with a different R group — same backbone, different chemistry
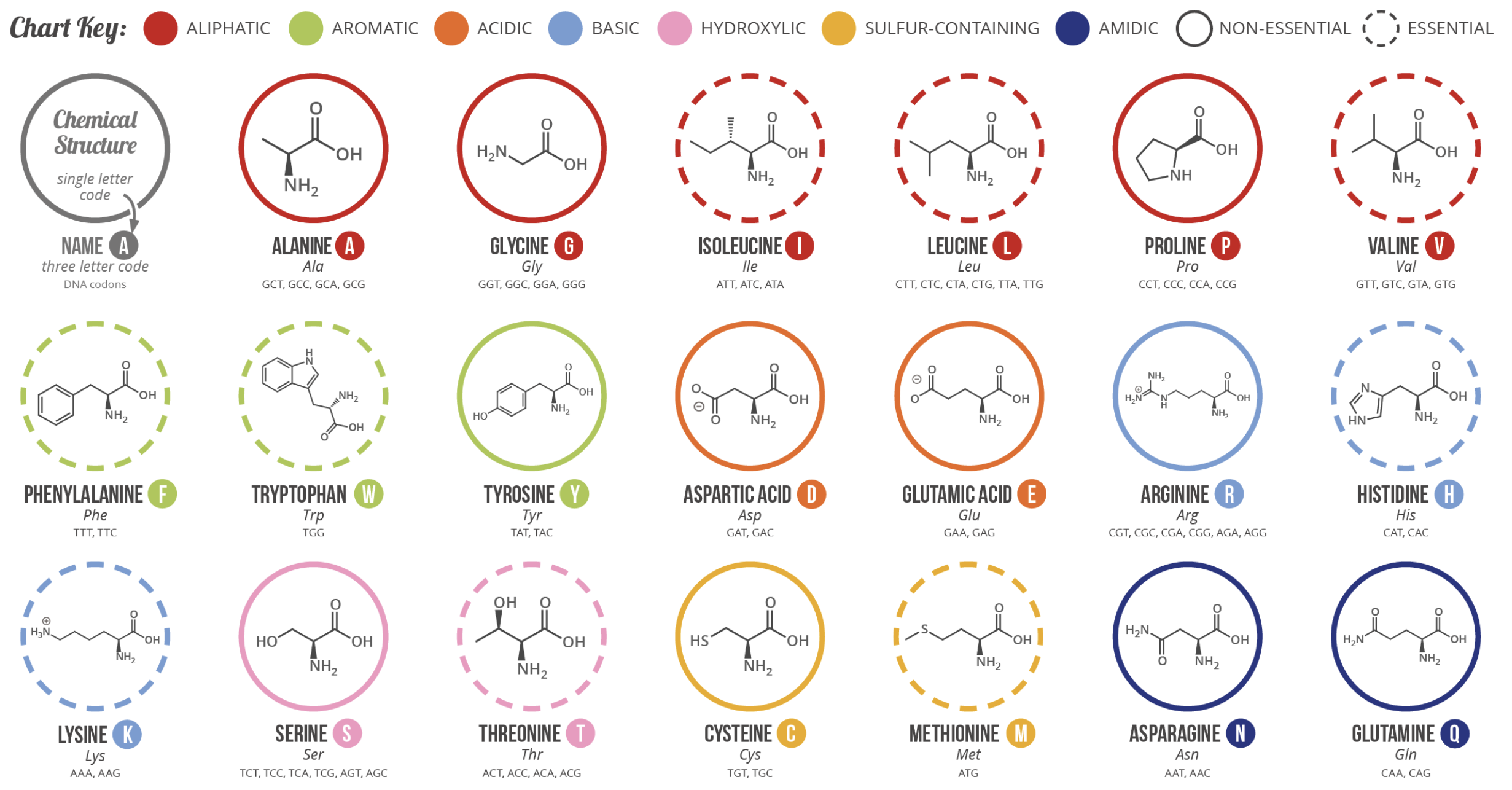
Primary Structure — The Amino Acid Sequence
- 20 standard amino acids, each with a unique side chain
- Typical protein: 300–500 amino acids
- Human genome encodes ~20,000 proteins
- Isoforms and post-translational modifications add many more
Amino Acids & the Protein Backbone
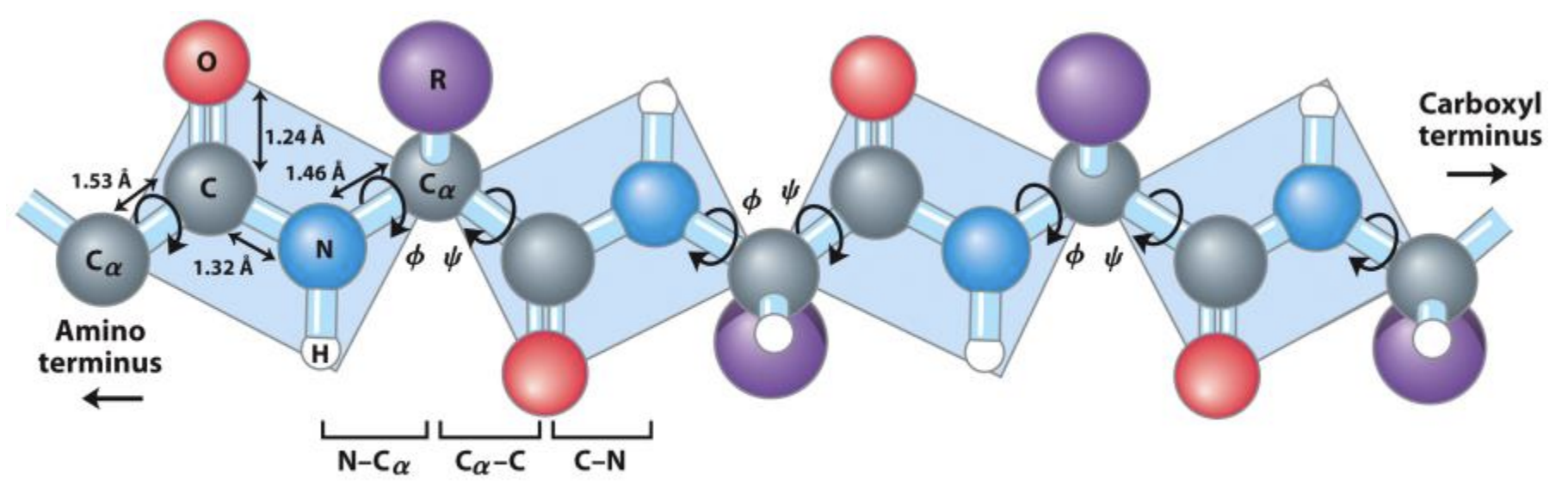
- All 20 amino acids share the same N–Cα–C backbone
- The R group (side chain) determines each amino acid's properties
- Amino acids link via peptide bonds
Secondary Structure
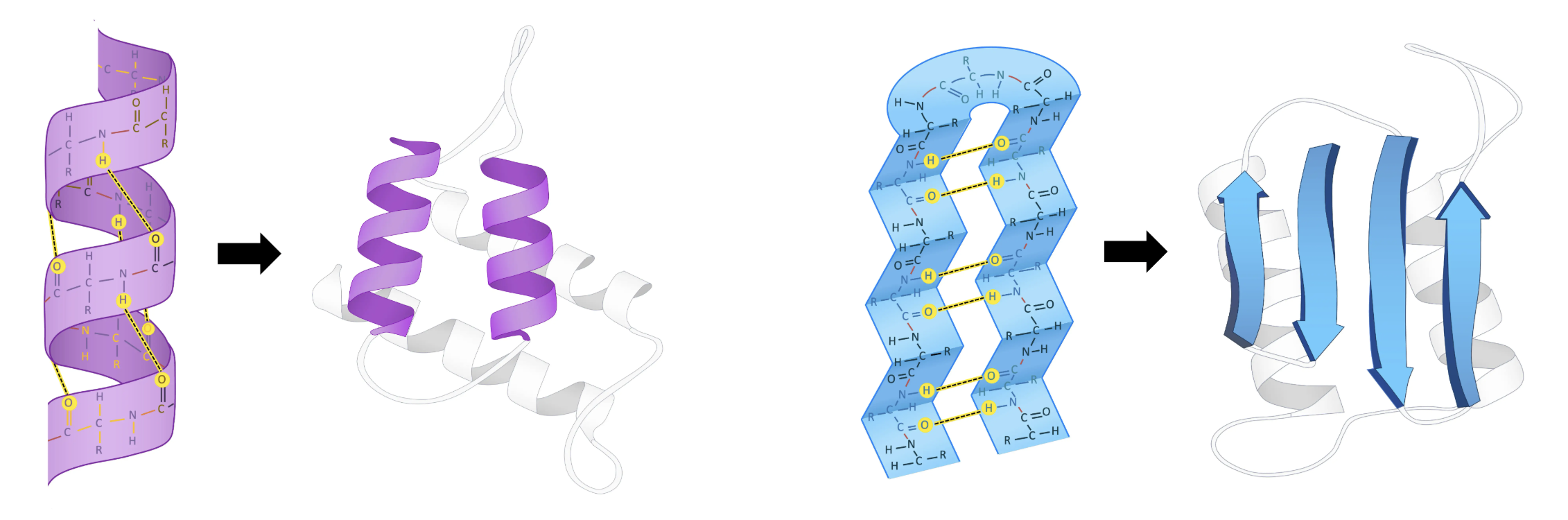
- Formed by hydrogen bonds between backbone atoms
- α-helices: coiled spring shape (~3.6 residues per turn)
- β-sheets: extended strands side by side (parallel or antiparallel)
Tertiary & Quaternary Structure
Tertiary
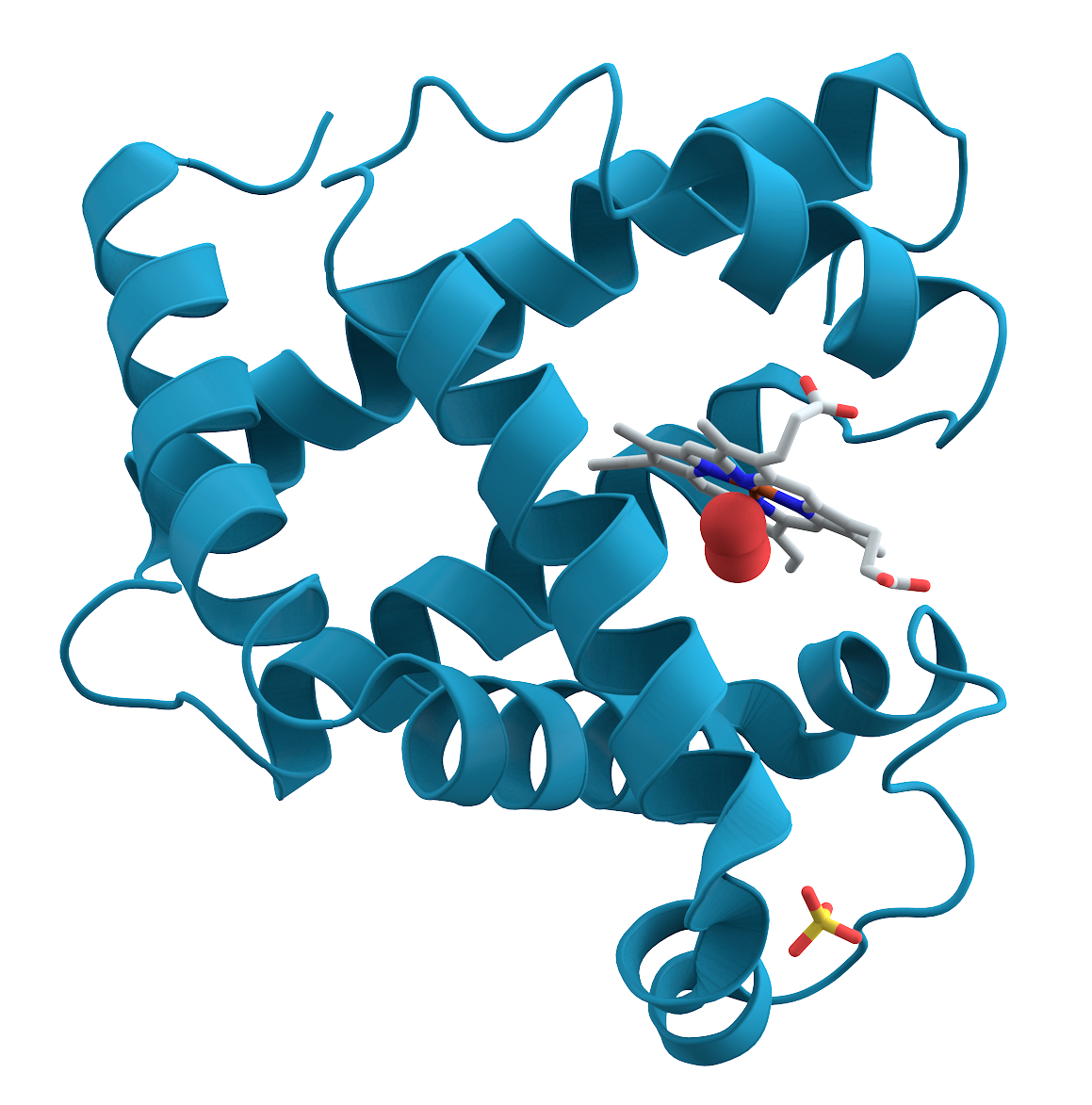
Quaternary
The Protein Data Bank
Founded 1971 with 7 structures — free, open access — key to every ML advance in structural biology
Without the PDB, there is no AlphaFold
How Are Protein Structures Determined?
X-ray Crystallography
~85% of PDB structures
Grow protein crystals, shoot X-rays, measure diffraction
Resolution: 1–3 Å
~25 Nobel Prizes
NMR Spectroscopy
~7% of PDB
Measures distances between atoms in solution
Limited to smaller proteins (<40 kDa)
7–8 Nobel Prizes
Cryo-Electron Microscopy
Revolution in the last decade
Flash-freeze proteins, image with electron beam
No crystals needed → larger complexes
1 Nobel Prize
Computational
AlphaFold & MD
2 Nobel Prizes
The PDB File Format
ATOM 1 N ALA A 1 27.340 24.430 2.614 1.00 9.67 N
ATOM 2 CA ALA A 1 26.266 25.413 2.842 1.00 10.38 C
ATOM 3 C ALA A 1 26.913 26.639 3.531 1.00 9.62 C
ATOM 4 O ALA A 1 27.886 26.463 4.263 1.00 9.62 O
ATOM 5 CB ALA A 1 25.112 24.880 3.649 1.00 13.77 C| Record | Serial | Name | Residue | Chain | ResSeq | X | Y | Z | Occ. | B-factor | Element |
| ATOM | 1 | N | ALA | A | 1 | 27.340 | 24.430 | 2.614 | 1.00 | 9.67 | N |
Every atom gets x, y, z coordinates in Ångströms (10−10 m)
Predicting Protein Structure
Can we predict 3D structure from sequence alone?
CASP — Critical Assessment of Protein Structure Prediction
Founded 1994 by John Moult
Biennial blind prediction competition
How it works:
- Experimentalists solve structures (not yet public)
- Predictors submit models before structures are released
- Models evaluated against experimental truth
Metric: GDT-TS
Global Distance Test — Total Score (0–100)
Fraction of residues within distance thresholds of the true structure
Truly prospective evaluation — no possible data leakage
The Arc of Protein Structure Prediction
AlphaFold 2 (2020)
Jumper et al., Nature 2021
- CASP14: GDT-TS ≈ 92.4 (experimental accuracy)
- Solved a 50-year grand challenge in biology
- Nobel Prize in Chemistry 2024 (Demis Hassabis, John Jumper)
AlphaFold: Data Engineering Matters
- Noisy student distillation
- PDB clustering at 40% identity — sample evenly across sequence clusters, then uniformly within clusters
- IPA replaced by data augmentation (AF3) — invariant point attention dropped in favor of random rotations/translations during training
To read more: The Illustrated AlphaFold by Elana Simon
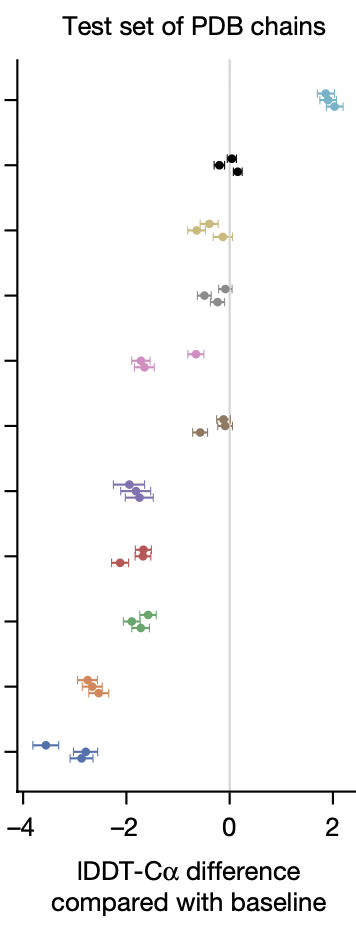
Impact of AlphaFold
- 200+ million structures predicted (AlphaFold Protein Structure Database)
- Covers nearly every known protein sequence
- Accelerated research across biology: drug design, enzyme engineering, evolutionary biology
But: AlphaFold predicts structure, not dynamics or function directly
And: it required the PDB — 50 years of painstaking experimental work
From Prediction to Design
Can we invert the process?
Instead of sequence → structure,
can we go function → structure → sequence?
RFdiffusion — Generative Protein Design
Watson et al., Nature 2023
- Uses denoising diffusion on protein backbones
- Based on RoseTTAFold (RF) architecture as denoising network
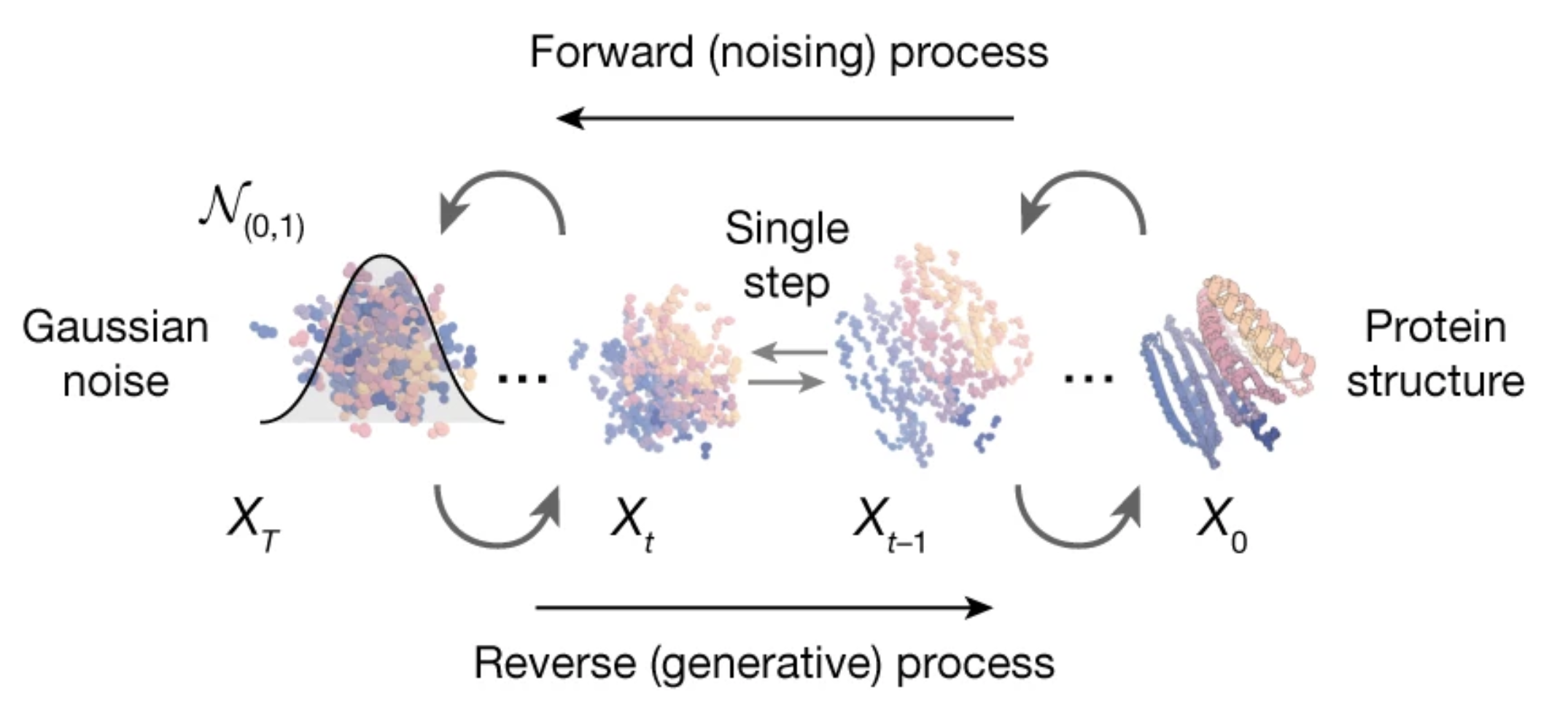
What Can RFdiffusion Design?
- Unconditional generation — entirely new protein folds
- Motif scaffolding — build a protein around a functional site
- Binder design — design proteins that bind to specific targets
- Symmetric assemblies — design symmetric protein complexes
- Partial diffusion — diversify existing designs
Experimentally validated: designed proteins fold correctly and bind targets as intended

Part II
Small Molecules
What Are Small Molecules?
Organic compounds, typically < 900 Da molecular weight
- Pharmaceuticals — ~90% of approved drugs are small molecules
- Agrochemicals (pesticides, herbicides, fungicides)
- Electronics (semiconductors, OLEDs)
- Climate (photovoltaics, battery electrolytes)
- Dyes and pigments
- Catalysts
- Cosmetics
- Industrial chemicals
- Explosives
Name That Molecule
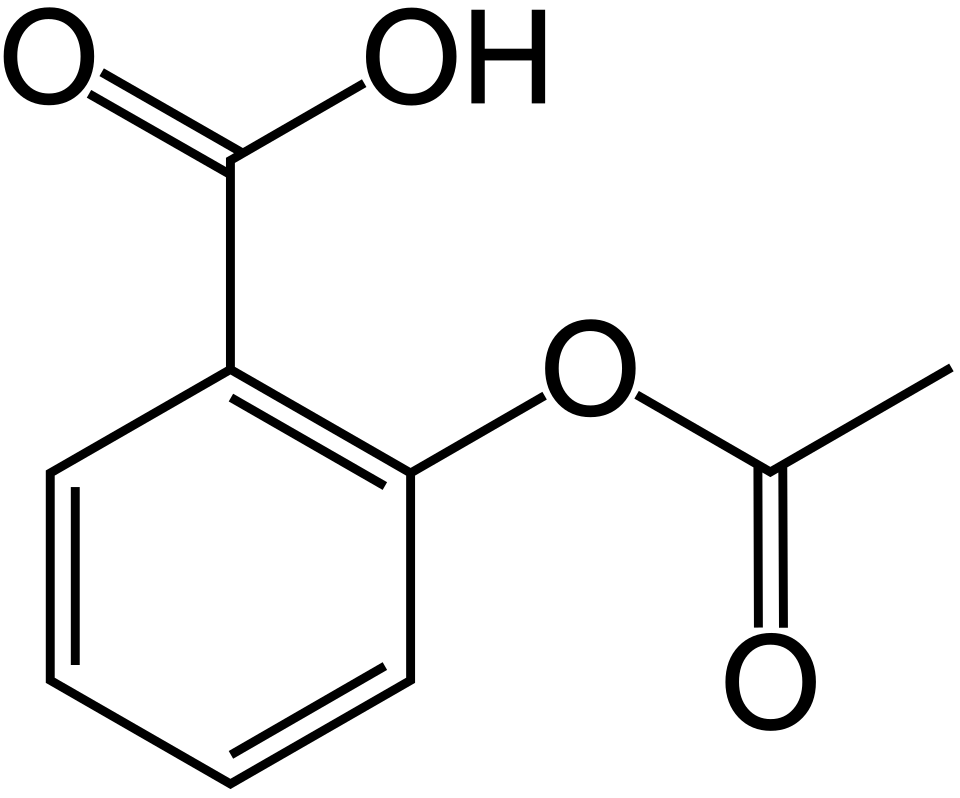
?
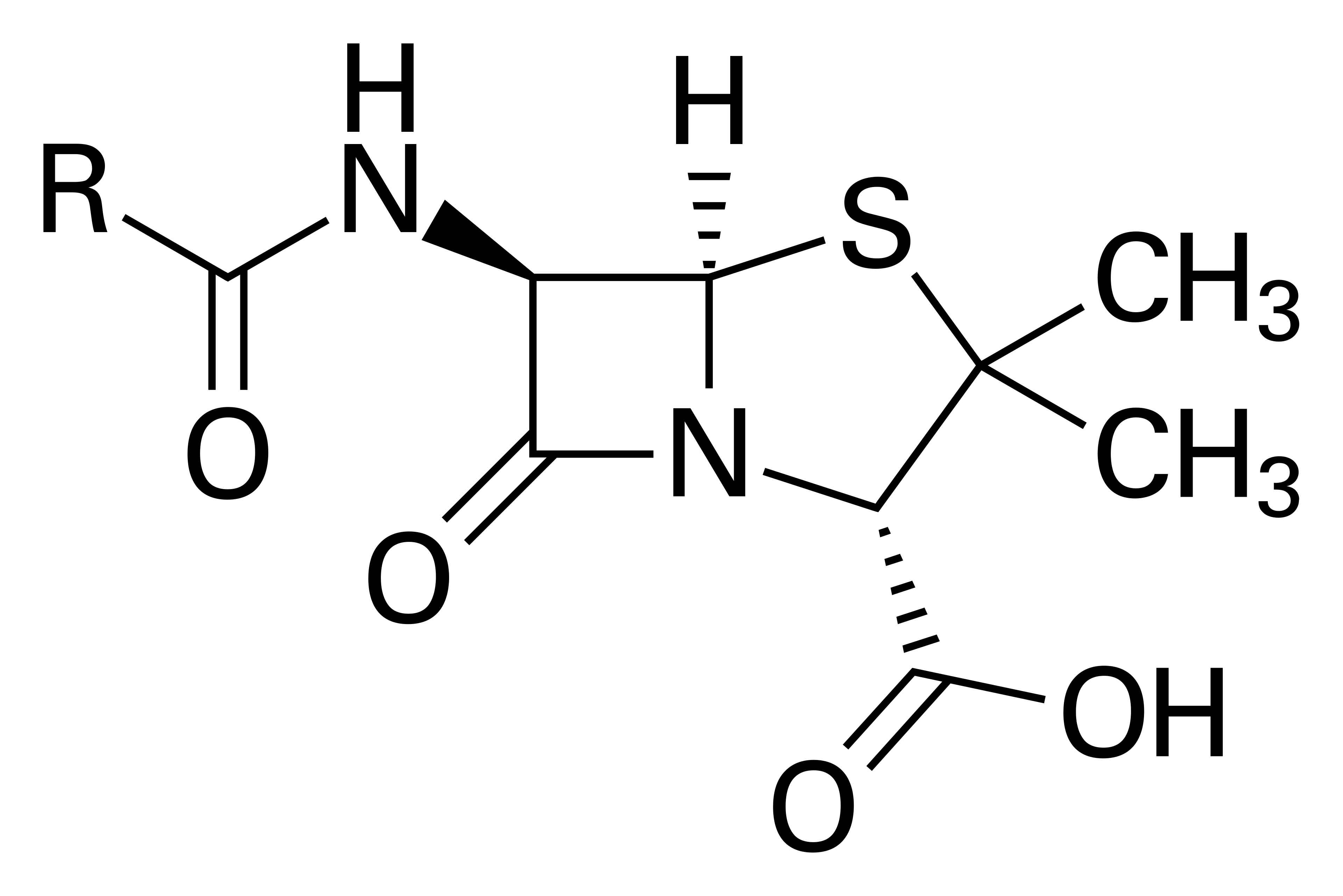
?
?
?
Name That Molecule
Aspirin
Penicillin G
Cholesterol
TNT
A Single Molecule Can Change the World
- Penicillin — saved an estimated 200 million lives
- Aspirin — used by 100+ million people daily
- Metformin — first-line diabetes treatment for 500M patients
- Atorvastatin (Lipitor) — $125 billion in total sales
- Artemisinin — Nobel Prize 2015, saves millions from malaria
Drug discovery is one of the highest-impact applications of ML
Quick Chemistry Refresher
Drug-like molecules are mostly C, H, N, O with some S, F, Cl
Bonds represent shared electron pairs
Single bond = 1 pair, double = 2, triple = 3
Aromatic rings: delocalized electrons shared across the ring
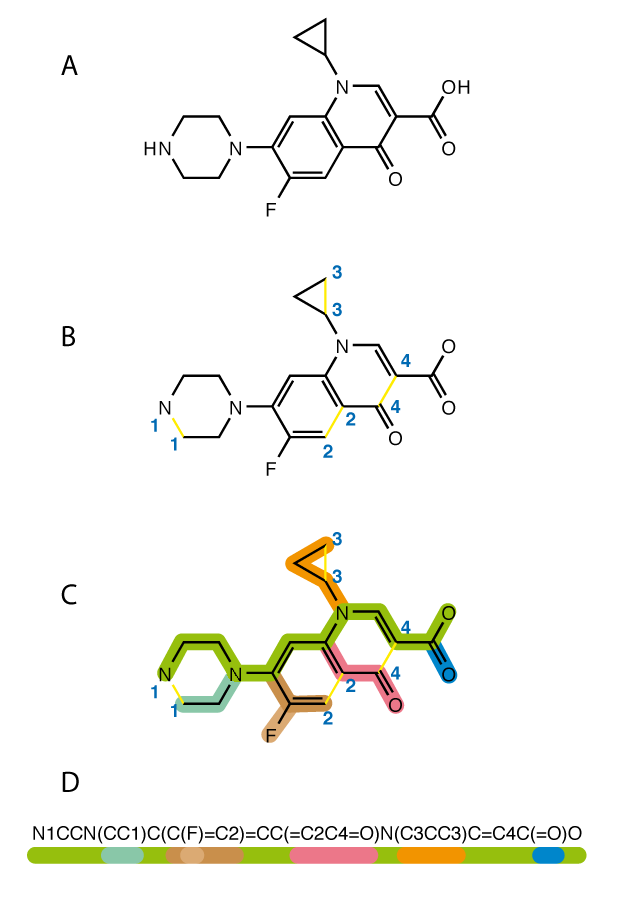
SMILES — Molecular Strings
Simplified Molecular-Input Line-Entry System (Weininger, 1988)
| Molecule | SMILES |
|---|---|
| Methane | C |
| Ethanol | CCO |
| Acetic acid | CC(=O)O |
| Benzene | c1ccccc1 |
| Aspirin | CC(=O)Oc1ccccc1C(=O)O |
| Caffeine | Cn1c(=O)c2c(ncn2C)n(C)c1=O |
Rules: atoms as letters, branches in (), rings by number pairs, lowercase = aromatic
Universal, compact, machine-readable representation
This is often the input format for ML models
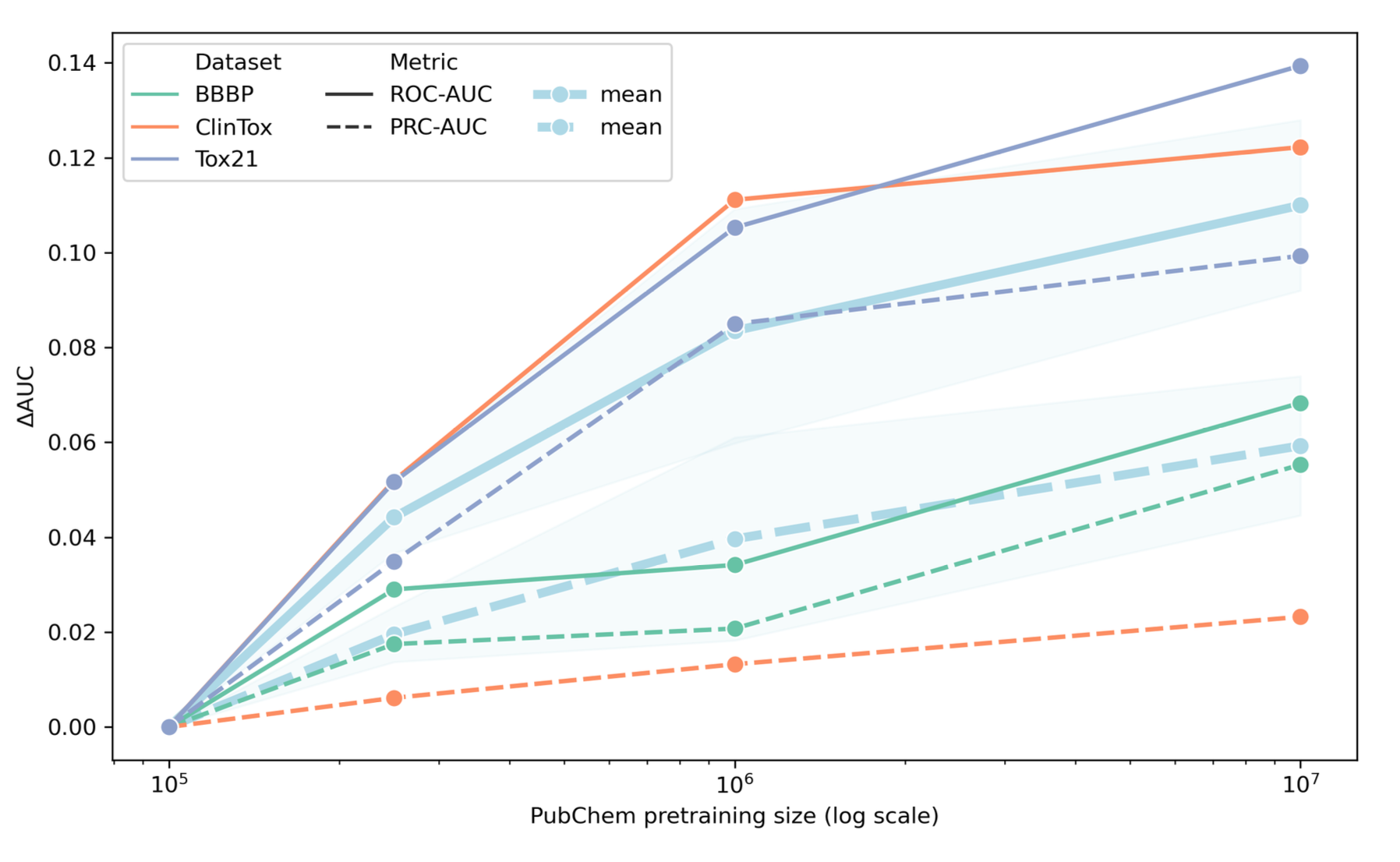
Why Is ML Harder for Small Molecules Than Proteins?
- Noisier labels — bioactivity data comes from heterogeneous assays with varying protocols; structure data is more uniform
- Graphs, not sequences — SMILES linearization is fragile (minor edits break validity), lacks natural tokenization
- Activity cliffs — a single atom change can shift potency by orders of magnitude; proteins have smoother fitness landscapes
- No evolutionary signal — proteins have hundreds of millions of evolved sequences for self-supervised learning; no equivalent for small molecules
- Tiny datasets, vast space — ~1060 drug-like molecules, but labeled datasets are small and inconsistent across sources
ECFP — Extended-Connectivity Fingerprints
Morgan (1965) → Rogers & Hahn (2010)
The dominant molecular representation in cheminformatics for decades
Each atom's local environment is encoded at increasing radii
Substructures are hashed to bits in a fixed-length vector (typically 1024 or 2048 bits)
Sparse, binary, interpretable — but not differentiable
Neural Graph Fingerprints
Convolutional Networks on Graphs for Learning Molecular Fingerprints
Duvenaud, Maclaurin, et al. — NeurIPS 2015 — 5,000+ citations
Key insight: Replace every non-differentiable operation in ECFP with a differentiable analog → end-to-end learning
| ECFP (Fixed) | Neural FP (Learned) |
|---|---|
| Hash function | Neural network layer |
| Concatenate neighbors | Sum neighbors |
| Index into bit vector | Softmax |
| Binary output | Real-valued output |
Neural Fingerprints — Results
| Method | Solubility | Drug Efficacy | Photovoltaic |
|---|---|---|---|
| Circular FPs + linear | 1.71 | 1.13 | 2.63 |
| Circular FPs + neural net | 1.40 | 1.36 | 2.00 |
| Neural FPs + linear | 0.77 | 1.15 | 2.58 |
| Neural FPs + neural net | 0.52 | 1.16 | 1.43 |
Values are RMSE — lower is better. Bold = best.
Neural fingerprints match or beat circular fingerprints on all tasks
This paper launched the field of graph neural networks for chemistry
ChemBERTa
Generative models for chemistry remain difficult
Protein–Ligand Binding Prediction
The Central Problem of Drug Discovery
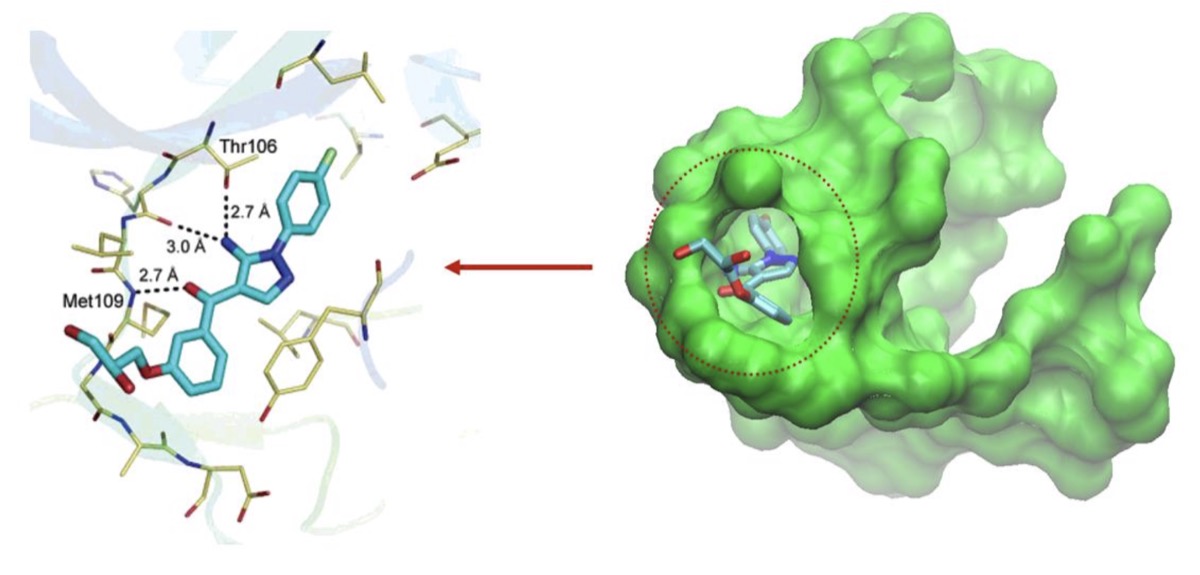
- Goal: find molecules that bind tightly and specifically to disease-relevant proteins
- ~$1B and ~10 years per approved drug
- Computational prediction: fast but challenging
Accurate binding prediction would revolutionize drug discovery
IC₅₀ vs Kᵢ vs Kd
IC₅₀
Concentration that inhibits 50% of activity
Depends on assay conditions (substrate conc, etc.)
Most commonly reported — but least comparable
Kᵢ
Inhibition constant
Corrected for substrate concentration (Cheng-Prusoff)
More comparable, but still assay-dependent
Kd
Dissociation constant
Thermodynamic quantity — measures true binding affinity
Gold standard, but hardest to measure
These values can differ by 10–100× for the same molecule–protein pair
The Data Quality Crisis
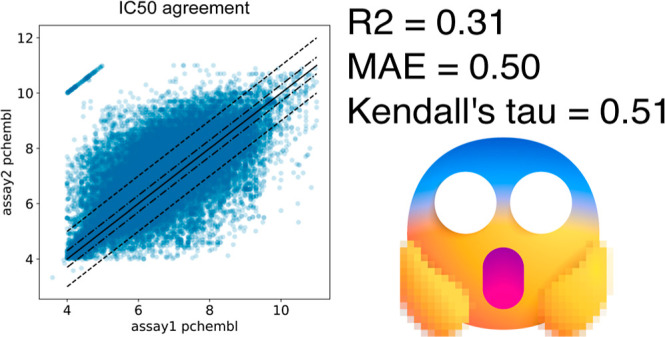
R² = 0.31 between IC₅₀ measurements from different sources
MAE = 0.50 log units (≈ 3× error)
Kalliokoski et al., 2013
If your labels have R²=0.31 with themselves, the ceiling for any model is R² ≈ 0.56 against those labels
Maximum achievable: R²max = σ²true / (σ²true + σ²exp) — Brown, Muchmore & Hajduk, Drug Disc. Today 2009
Why Binding Prediction Is Hard
- Noisy data
- Positivity bias — Datasets massively overrepresent known binders; true negatives are unknown
- Pocket overfitting — Models memorize specific protein binding sites rather than learning general binding physics
- Ligand-only signal — Often, most predictive power comes from the ligand alone (its size, lipophilicity) without any protein information
- Conformation — Both protein and ligand change shape upon binding (induced fit)
Many published models are no better than memorizing the training set — careful evaluation is critical
Boltz-2 — Structure + Affinity Prediction
Passaro et al., 2025 — Fully open source
Predicts both 3D complex structure AND binding affinity
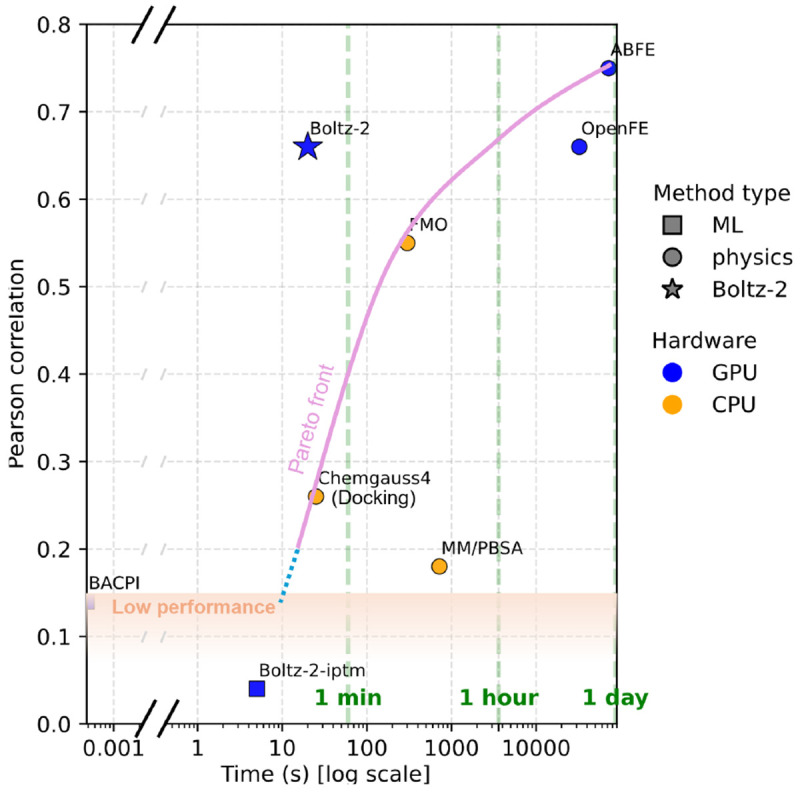
- Near ABFE accuracy — 1000× faster
- Open weights, open training code
- Input: protein sequence + ligand as molecular graph
- Trained on PDB structures + ChEMBL binding data
AlphaFold 3 — Cofolding Everything
Abramson et al., Nature 2024
Predicts structures of proteins + nucleic acids + small molecules + ions + modified residues — all together
Key advances over AF2:
- Diffusion-based structure module (instead of equivariant point attention)
- Small molecules, DNA, RNA, ions as first-class inputs
- Single model for all biomolecular complexes
This is cofolding: predicting how everything folds and binds together
Is AI for Chemistry the Overfitting Olympics?
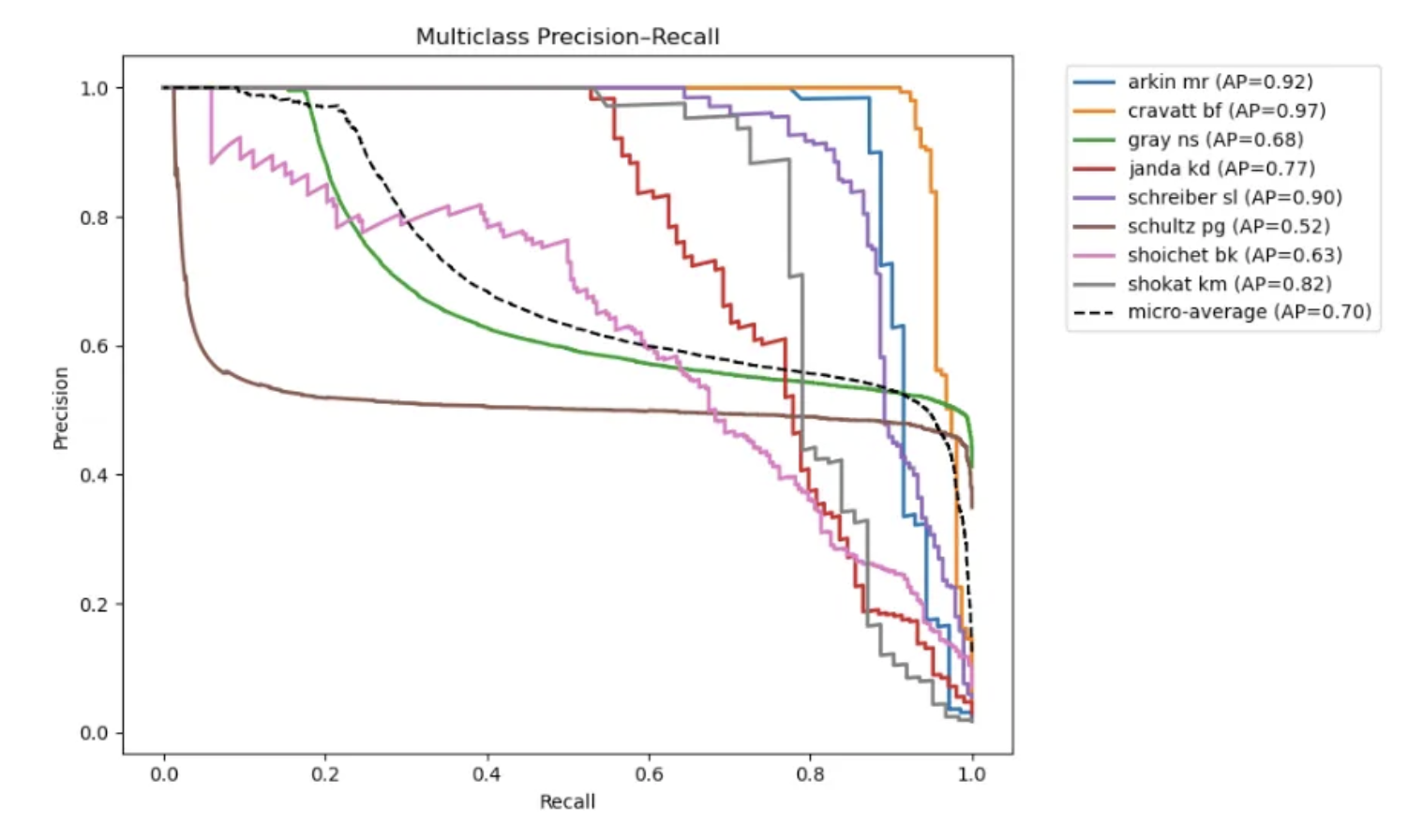
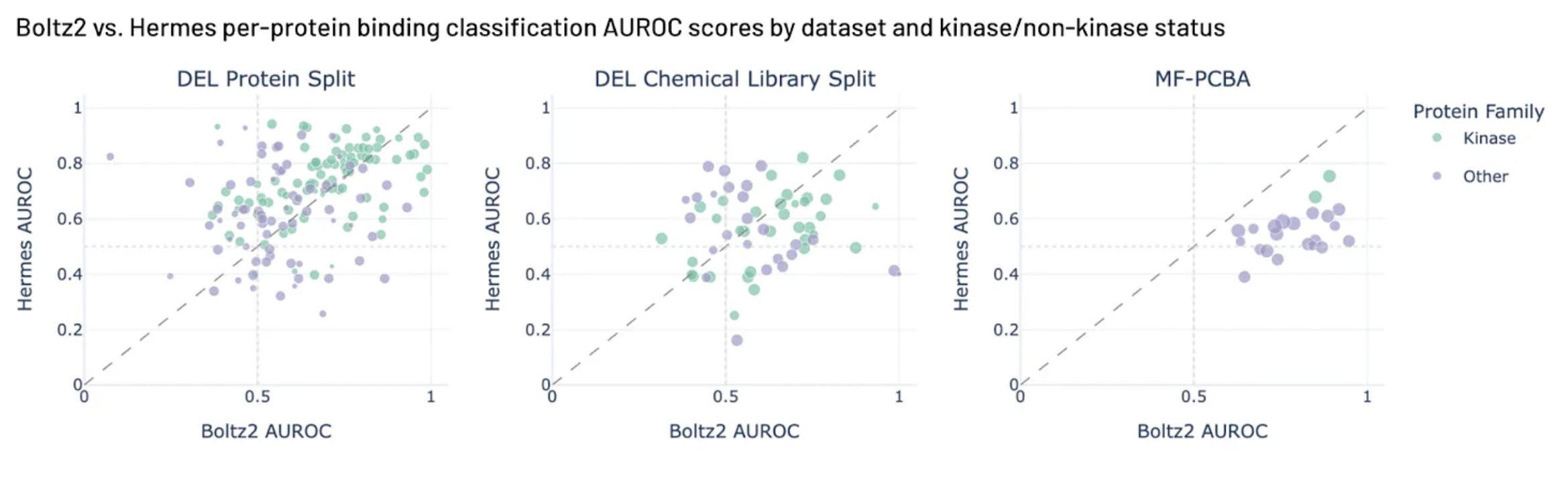
Maybe It's the Data, Not the Architecture
SimpleFold
Jing et al., 2025 — arXiv:2509.18480
- Much simpler model than AlphaFold 2
- ~800 lines of Python for core training code
- ~25× faster forward pass vs. AF2
- Distilled on AF2's predicted structures — not trained on PDB
- Approaches AF2-level accuracy
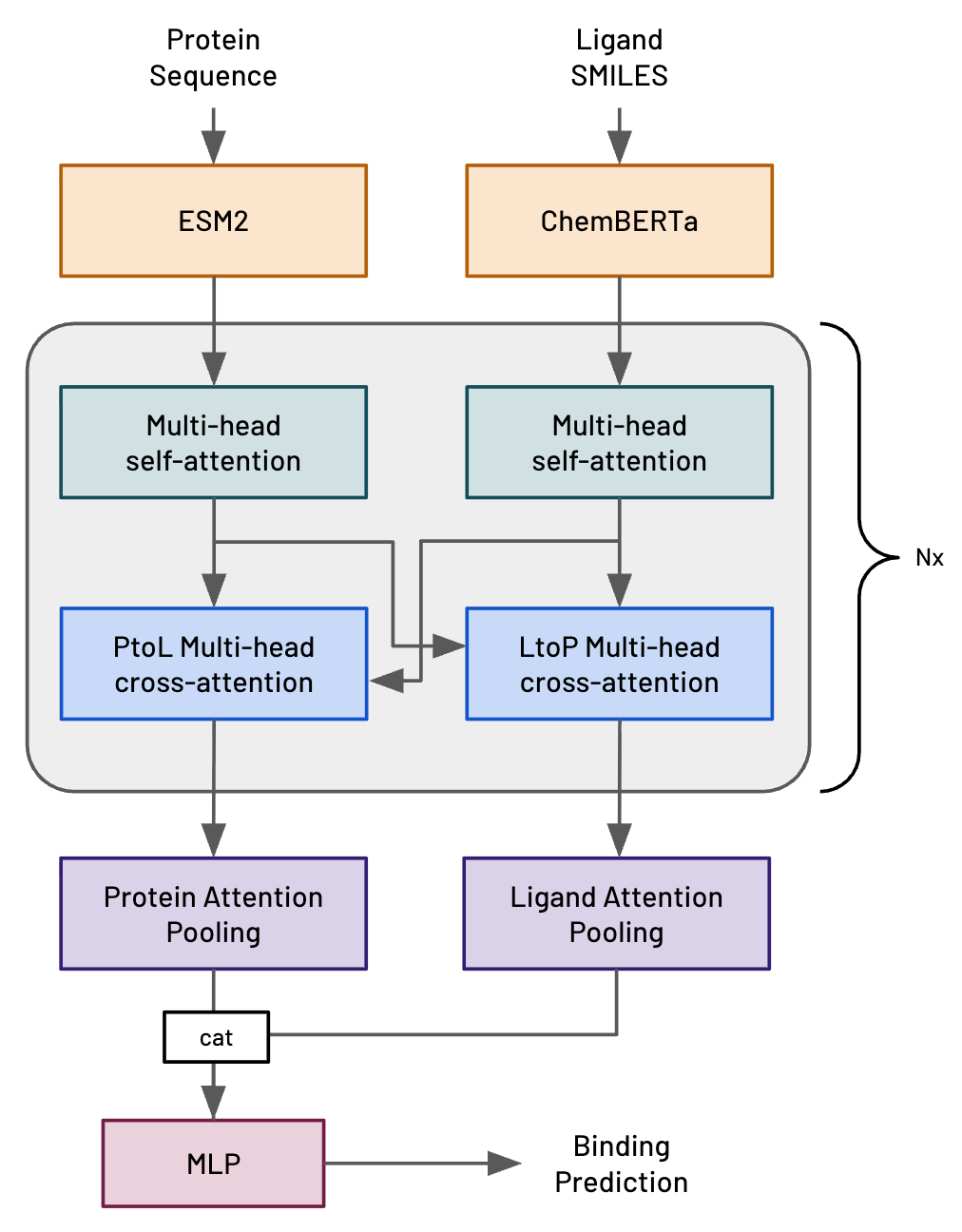
HERMES (Leash Bio)
- Simple cross-attention architecture (ESM2 + ChemBERTa)
- Trained on massive DEL screening data from their own platform
- Competitive with Boltz-2 on binding prediction
Key Takeaways
- A simple model trained on great data will beat a complex model trained on bad data
- Data + evaluation → breakthroughs: the PDB and CASP enabled AlphaFold; ChEMBL and PDBBind enabled binding prediction
- Representation matters: sequences, backbone atoms, 3D coordinates, SMILES, fingerprints, graphs
- Binding prediction is hard: noisy labels, biased data, evaluation pitfalls
- The open question: do sophisticated biophysical methods (AF3, Boltz-2) beat simple models trained on tons of data — or is there a best of both worlds?
Appendix
The Kinetochore
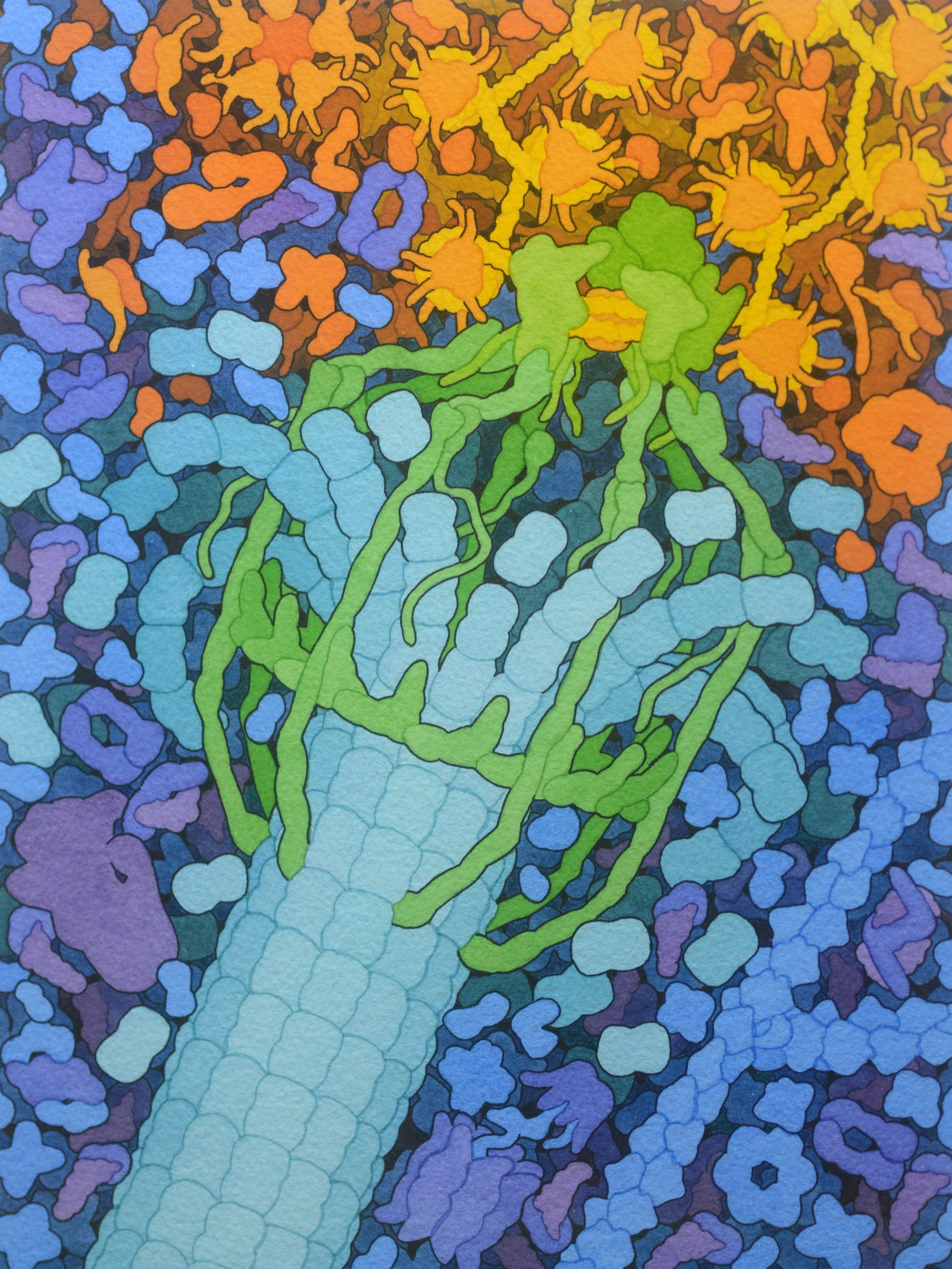
Connects chromosomes to spindle fibers during cell division
A complex of ~100 different proteins working together
Image: David S. Goodsell, RCSB PDB
From Hash Functions to Neural Networks
Left: Molecular graph → message passing between atoms → pooling → fingerprint
At each layer, each atom aggregates information from its neighbors
Identical to ECFP with large random weights — but now we can optimize
Interpretable Learned Features
Feature predictive of solubility
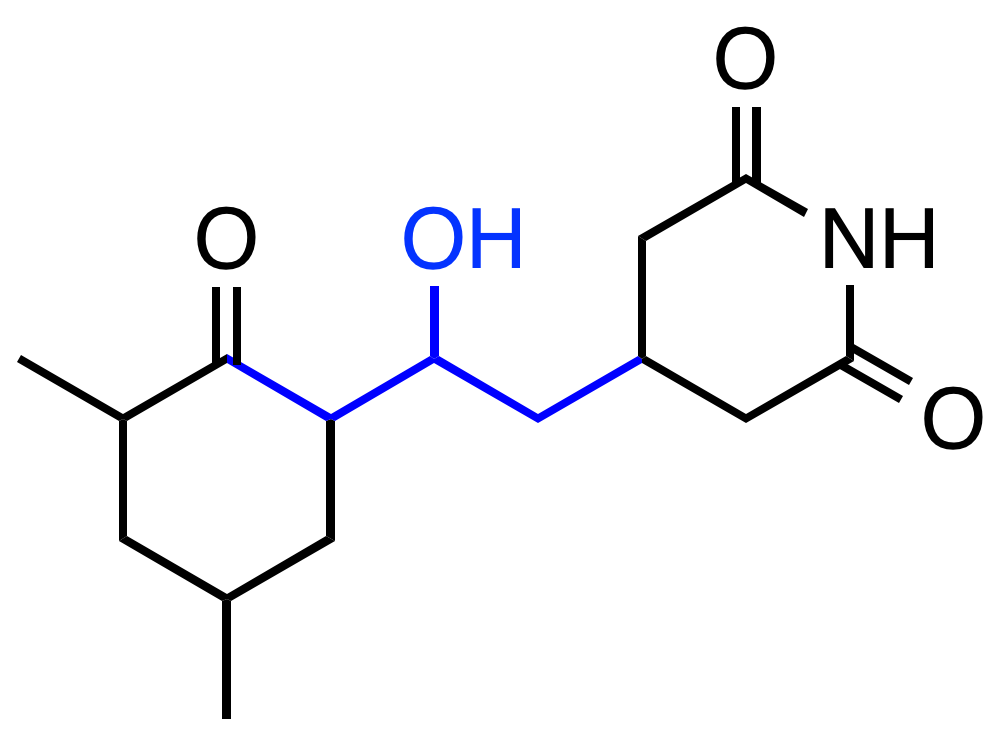
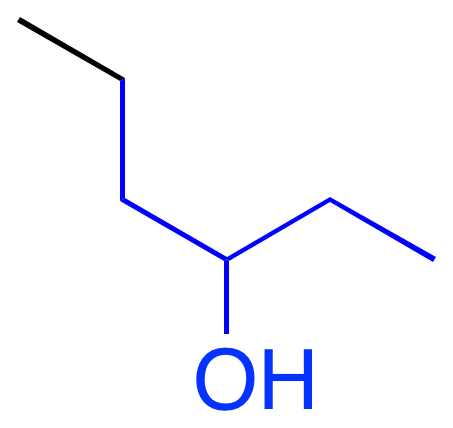
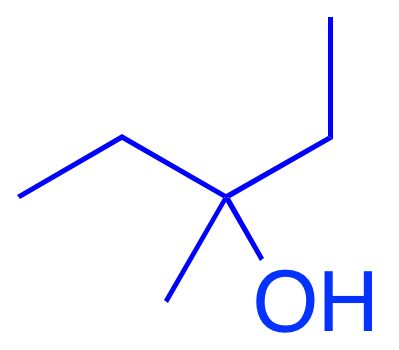
Pro-solubility features activate on hydrophilic OH groups
Feature predictive of toxicity
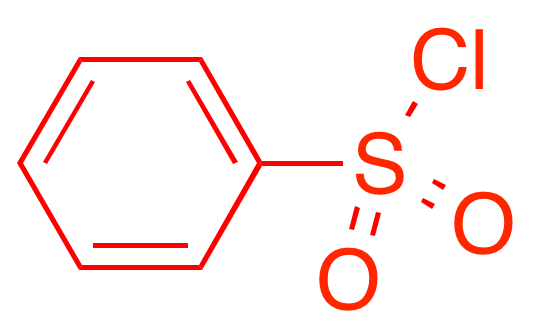
Pro-toxicity features activate on aromatic ring systems (known carcinogens)
Unlike ECFP, learned features can be activated by similar but distinct fragments
The Recipe for a Breakthrough
A large, high-quality dataset
The Protein Data Bank (50 years of data)
A standardized metric
GDT-TS score
A truly prospective competition
CASP (no data leakage possible)
This combination is rare in biology — and it enabled one of the most important ML breakthroughs in history
AlphaFold 3 — Full Architecture
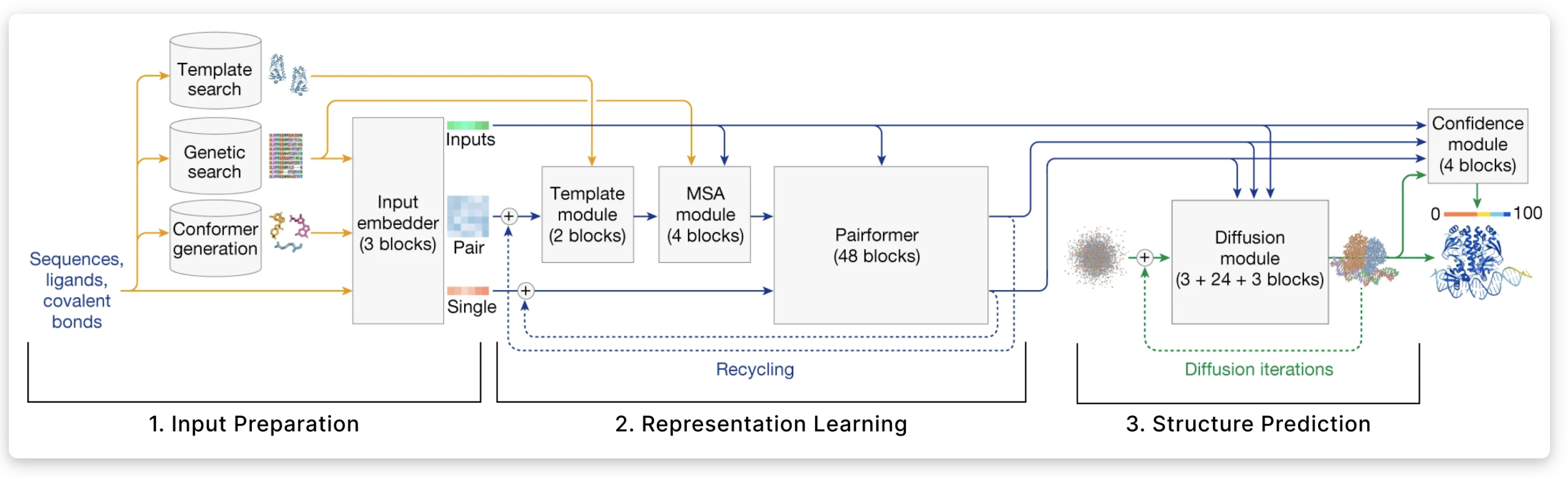
AlphaFold 2 — Architecture Overview
Why AlphaFold 2 Worked
- Multiple Sequence Alignments (MSAs) — evolutionary information from related proteins
- Evoformer — attention mechanism that reasons about residue pairs
- Structure Module — directly predicts 3D coordinates using equivariant transformations
- End-to-end differentiable — trained on PDB structures with FAPE loss
- Recycling — iteratively refines its own predictions
Maximum Achievable R² Under Label Noise
Brown, Muchmore & Hajduk, Drug Discovery Today 2009
Two independent measurements of true value \(T\): \(\;X_1 = T + \varepsilon_1,\; X_2 = T + \varepsilon_2,\; \varepsilon \sim \mathcal{N}(0, \sigma^2_{\text{exp}})\)
$$r(X_1, X_2) = \frac{\sigma^2_{\text{true}}}{\sigma^2_{\text{true}} + \sigma^2_{\text{exp}}} = \rho \quad \Rightarrow \quad R^2(X_1, X_2) = \rho^2$$
For a perfect model \(M = T\) evaluated against noisy labels:
$$R^2_{\max} = \rho = \sqrt{R^2(X_1, X_2)}$$
Intuition: Two noisy measurements → noise on both sides → R² = 0.31
Perfect model vs noisy labels → noise on one side only → R² = 0.56
Example: R²(X₁, X₂) = 0.31 → R²max = √0.31 ≈ 0.56 — if your model hits ~0.5, more capacity won't help; you need better labels
Phi (φ) and Psi (ψ) Angles
- Each residue is described by two torsion angles: φ and ψ
- The peptide bond (ω) is nearly always planar (trans)
- φ, ψ define the local backbone conformation
The Ramachandran Plot
- Steric clashes restrict φ, ψ to specific regions
- Powerful validation tool: ~98% of residues should fall in allowed regions
- A compact, local representation of protein structure
β-sheet: φ ≈ −120°, ψ ≈ 130°
α-helix: φ ≈ −60°, ψ ≈ −45°
L-helix: φ ≈ 60°, ψ ≈ 45°